

Bishydrazide to Synthesize oxadiazoles

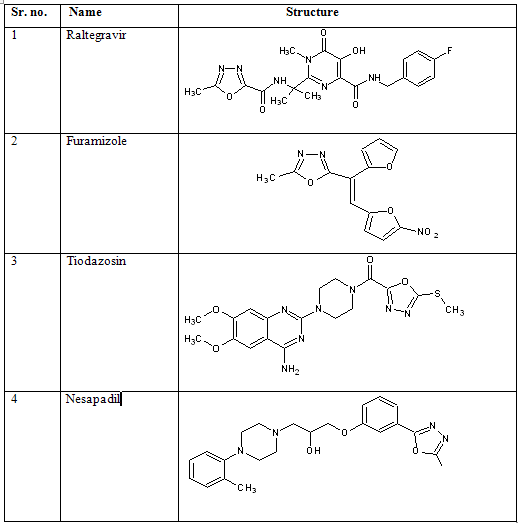
Drugs containing 1, 3, 4-oxadiazole moiety
http://www.pharmatutor.org/articles/Chemistry-common-synthetic-route-oxadiazole-%20important-heterocyclic-moiety-in-medicinal-chemistry?page=0,5
http://www.pharmatutor.org/articles/Chemistry-common-synthetic-route-oxadiazole-%20important-heterocyclic-moiety-in-medicinal-chemistry?page=0,0



Novel 2-amino-5-substituted-1,3,4-oxadiazoles have been synthesized in excellent yields using the synthetic route outlined in Scheme 1. IR, 1H NMR, 13C NMR and mass spectral data are in agreement with the proposed structures of all synthesized compounds.
Lack of 1H NMR resonances observed with NH and NH2 functions in the1H NMR spectrum of 1a-n proved that ring closure starting from 4 resulted in the formation of 2-amino-1,3,4-oxadiazole ring.
This was further substantiated by the 13C NMR data of 1 which showed the peaks at 8 170-173 and 145-150 due to C2 and C5 of oxadiazole respectively. The IR spectrum shows 1600-1620 ern1 for (C=N-N=C) and 1063-1073 cm-1 for (C-O-C) in the compounds 1a-1n which confirmed the synthesis of 1,3,4-oxadiazoles.
http://www.scielo.cl/scielo.php?pid=S0717-97072010000100030&script=sci_arttext
1,3,4-oxadiazole is a versatile lead molecule for designing potential bioactive agents. The 1,3,4-oxadiazole derivatives have been found to exhibit diverse biological activities such as hypotensive1 anti-microbial2-4, anti-HIV2-4, anti-fungal5-6 anti-inflammatory7 antimitotic activity8 and muscle relaxant9
It have been found in the líterature study that the methods for synthesis of oxadiazole 1 include bromine oxidation of semicarbazide derivative and the cyclodesulfurization of acylthiosemicarbazide derivatives in the solution using I2/NaOH or 1,3-dicyclohexylcarbodimide (DCC)10-13 as well as mercury(II) acetate (Hg(OAc)2) or yellow mercury(II) oxide HgO14-16 Evans17 synthesized oxadiazole derivatives by rapid parallel synthesis in efficient one-pot preparation using resin-bound reagents. All these methods are usually carried out in various different synthetic steps and require the heating at higher temperature. The handling of these reagents is not only difficult but also very hazardous to environment. The each stage of the reaction including extraction and purification of the producís from the mixture requires great precautions.

EXAMPLE 1 3-(Trifluoromethyl -5,6,7,8-tetrahydrori,2,41triazolor4.3-α1pyrazine, hydrochloride salt (1-4)
Step A: Preparation of bishydrazide (1-1)

H
Hydrazine (20.1 g, 35 wt% in water, 0.22 mol) was mixed with 310 mL of acetonitrile. 31.5 g of ethyl trifluoroacetate (0.22 mol) was added over 60 min. The internal temperature was increased to 25 °C from 14 °C. The resulting solution was aged at 22 - 25 °C for 60 min. The solution was cooled to 7 °C. 17.9 g of 50 wt% aqueous NaOH (0.22 mol) and 25.3 g of chloroacetyl chloride (0.22 mol) were added simultaneously over 130 min at a temperature below 16 °C. When the reaction was complete, the mixture was vacuum distilled to remove water and ethanol at 27 ~ 30 °C and under 26 ~ 27 in Hg vacuum. During the distillation, 720 mL of acetonitrile was added slowly to maintain constant volume (approximately 500 mL). The slurry was filtered to remove sodium chloride. The cake was rinsed with about 100 mL of acetonitrile. Removal of the solvent afforded bis-hydrazide \Λ
(43.2 g, 96.5% yield, 94.4 area% pure by HPLC assay).
1H-NMR (400 MHz, DMSO-dfc): δ 4.2 (s, 2H), 10.7 (s, 1H), and 11.6 (s, 1H) ppm.
13C-NMR (125 MHz, DMSO-_f6): δ 41.0, 116.1 (q, J = 362 Hz), 155.8 (q, J = 50 Hz), and 165.4 ppm.
HPLC conditions: Symmetry 4.6 x 250 mm C18 column; UV detection at 210 nm; mobile phase: 1:1 ACN: H2O (0.1% H3PO ); flow rate: 1 rnL/min; retention time of
1-1: 2.9 min.
Step B: Preparation of 5-(trifluoromethyl)-2-(chloromethyl)- 1.3 ,4-oxadiazole
F3C
Bishydrazide LI from Step A (43.2 g, 0.21 mol) in ACN (82 mL) was cooled to
5 °C. Phosphorus oxychloride (32.2 g, 0.21 mol) was added, maintaining the temperature below 10 °C. The mixture was heated to 80 °C and aged at this temperature for 24 h until HPLC showed less than 2 area% of LI. In a separate vessel, 260 mL of LPAc and 250 mL of water were mixed and cooled to 0 °C. The reaction slurry was charged to the quench keeping the internal temperature below 10 °C. After the addition, the mixture was agitated vigorously for 30 min, the temperature was increased to room temperature and the aqueous layer was cut. The organic layer was then washed with 215 mL of water, 215 mL of 5 wt% aqueous sodium bicarbonate and finally 215 mL of 20 wt% aqueous brine solution. HPLC assay yield after work up was 86-92%. Volatiles were removed by distillation at 75-80 mm Hg, 55 °C to afford an oil which could be used directly in Step C without further purification. Otherwise the product can be purified by distillation to afford L2 in 70-80% yield. 1H-NMR (400 MHz, CDC13): 04.8 (s, 2H) ppm.
13C-NMR (125 MHz, CDC13): δ 32.1, 115.8 (q, J = 337 Hz), 156.2 (q, J = 50 Hz), and 164.4 ppm. HPLC conditions: Symmetry 4.6 x 250 mm C18 column; UV detection at 210 nm; mobile phase: 1:1 ACN: H2O (0.1% H3PO4); flow rate: 1 rnL/min; retention time of 1-2: 8.8 min.

 Synthesis of 1,3,4-oxadiazoles
Synthesis of 1,3,4-oxadiazoles
Recent Literature

A direct access to symmetrical and unsymmetrical 2,5-disubstituted [1,3,4]-oxadiazoles has been accomplished through an imine C-H functionalization of N-arylidenearoylhydrazide using a catalytic quantity of Cu(OTf)2. These reactions can be performed in air atmosphere and moisture making it exceptionally practical for application in organic synthesis.
S. Guin, T. Ghosh, S. K. Rout, A. Banerjee, B. K. Patel, Org. Lett., 2011, 13, 5976-5979.

A facile and general protocol for the preparation of 2-amino-1,3,4-oxadiazoles relies on a tosyl chloride/pyridine-mediated cyclization of thiosemicarbazides that consistently outperforms the analogous semicarbazide cyclizations. Various 5-alkyl- and 5-aryl-2-amino-1,3,4-oxadiazoles have been prepared in good yields.
S. J. Dolman, F. Gosselin, P. D. O'Shea, I. W. Davies, J. Org. Chem., 2006, 71, 9548-9551.

An oxidative desulfurization approach enables the construction of oxadiazole and thiadiazole heterocycles in the presence of iodobenzene and Oxone. The use of iodobenzene and the inexpensive readily available oxidant Oxone makes the reaction system simple and versatile for desulfurization.
K. N. Patel, N. C. Jadhav, P. B. Jagadhane, V. N. Telvekar, Synlett, 2012, 23, 1970-1972.

The reaction of a thiosemicarbazide intermediate with EDC·HCl in DMSO or p-TsCl, triethylamine in N-methyl-2-pyrrolidone gives the corresponding 2-amino-1,3,4-oxadiazoles and 2-amino-1,3,4-thiadiazoles through regioselcective cyclization processes.
S.-J. Yang, S.-H. Lee, H.-J. Kwak, Y.-D. Gong, J. Org. Chem., 2013, 78, 438-444.

N-Isocyaniminotriphenylphosphorane, aldehydes, and benzoic acids undergo a one-pot, three-component reaction under mild conditions to afford 2-aryl-5-hydroxyalkyl-1,3,4-oxadiazoles in good yields.
M. Adib, M. R. Kesheh, S. Ansari, H. R. Bijanzadeh, Synlett, 2009, 1575-1578.
Symmetric and unsymmetric 1,3,4-oxadiazoles were synthesized in situ from hydrazine hydrate and the corresponding 2-acyl-4,5-dichloropyridazin-3-ones as acylating agents in polyphosphoric acid (PPA) or BF3·OEt2 in excellent yields.
Y.-D. Park, J.-J. Kim, H.-A. Chung, D.-H. Kweon, S.-D. Cho, S.-G. Lee, Y.-J. Yoon, Synthesis, 2003, 560-564.

A simple and straightforward method for the direct carboxylation of aromatic heterocylces such as oxazoles, thiazoles, and oxadiazoles using CO2 as the C1 source requires no metal catalyst and only Cs2CO3 as the base. A good functional group tolerance is achieved.
O. Vechorkin, N. Hirt, X. Hu, Org. Lett., 2010, 12, 3567-3569.

REFERENCES
1. M. Tyagi, A. Kumar, Oriental. J. Chem., 18, 125, (2002). [ Links ]
2. B. S. Holla, R. Gonaslaves, S. Shenoy, Eur. J. Med. Chem., 35, 267, (2000). [ Links ]
3. N. Cesur, S. Birteksoz, G. Otuk, Acta. Pharm. Turcica, 44, 23, (2002). [ Links ]
4. U. V. Laddi, S. R. Desai, R. S. Bennur, S. C. Bennur, Iridian J. Heterocycl. Chem., 11, 319, (2002). [ Links ]
5. X. Zou, Z. Zhang, G. Jin, J. Chem. Res. Synopses., 228 (2002). [ Links ]
6. X. J. Zou, L. H. Lai, G. Y. Jin, Z. X. Zhang, J. Agric. Food Chem., 50, 3757, (2002). [ Links ]
7. E. Palaska, G. Sohin, P. Kclicen, N. T. Darlu, G. Altinok, Farmaco, 57, 101 (2002). [ Links ]
8. A. Afiatpour, R. M. Srivastava, M. L. Oliveira, E. J. Barreiro, Braz. J. Med. Biol. Res., 27, 1403, (1994). [ Links ]
9. L. B. Clapp, A. R. Katritzky, C. W. Rees Eds., Comprehensive Heterocyclic Chemistry, Pergamon Press, Oxford, 1984. [ Links ]
10. R. S. Gani, S. S. Pujar, G. S. Gadaginamath, Indian J. Heterocycl. Chem., 12, 25, (2002). [ Links ]
11. S. M. Golovlyova, Y. A. Moskvichey, E. M. Alov, D. B. Kobylinsley, V. V. Ermolaeva, Chem. Heterocycl. Compd., 37, 1102, (2001). [ Links ]
12. F. M. Liu, B. L. Wang, Z. F. Zhang, Youji Huaxue, 21, 1126, (2001). [ Links ]
13. O. M. Aboulwafa, A. M. Ornar, Sulfur Lett., 14, 181, (1992). [ Links ]
14. H. M. Faidallah, E. M. Sharshira, S. A. Basaif, A. E. A-Ba-Oum, Phos. Sulf. Sil. Rel. Elem., 67, 177, (2002). [ Links ]
15. A. Hetzheim, K. Moeckel, Adv. Heterocyclic Chem., 07, 183, (1966). [ Links ]
16. J. Hill, In: K.T. Potts Eds., Comprehensive Heterocyclic Chemistry, Pergamon Press, Oxford, 06, 427, (1984). [ Links ]
17. F. T. Cappo, K. A. Evans, T. L. Graybill, G. Burton, Tetrahedron Lett., 45, 3257, (2004). [ Links ]



 DR ANTHONY MELVIN CRASTO Ph.D , Born in Mumbai in 1964 and graduated from Mumbai University, Completed his PhD from ICT ,1991, Mumbai, India in Organic chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with GLENMARK- GENERICS LTD, Research centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Prior to joining Glenmark, he worked with major multinationals like Hoechst Marion Roussel, now Sanofi Aventis, & Searle India ltd, now Rpg lifesciences, etc. He has worked in Basic research, Neutraceuticals, Natural products, Flavors, Fragrances, Pheromones, Vet Drugs, Drugs, formulation, GMP etc. He has total 25 yrs exp in this field, he is now helping millions, has million hits on google on all organic chemistry websites.His New Drug Approvals , Green Chemistry International, Eurekamoments in Organic Chemistry , WIX BLOG , WORLD DRUG TRACKER
DR ANTHONY MELVIN CRASTO Ph.D , Born in Mumbai in 1964 and graduated from Mumbai University, Completed his PhD from ICT ,1991, Mumbai, India in Organic chemistry, The thesis topic was Synthesis of Novel Pyrethroid Analogues, Currently he is working with GLENMARK- GENERICS LTD, Research centre as Principal Scientist, Process Research (bulk actives) at Mahape, Navi Mumbai, India. Prior to joining Glenmark, he worked with major multinationals like Hoechst Marion Roussel, now Sanofi Aventis, & Searle India ltd, now Rpg lifesciences, etc. He has worked in Basic research, Neutraceuticals, Natural products, Flavors, Fragrances, Pheromones, Vet Drugs, Drugs, formulation, GMP etc. He has total 25 yrs exp in this field, he is now helping millions, has million hits on google on all organic chemistry websites.His New Drug Approvals , Green Chemistry International, Eurekamoments in Organic Chemistry , WIX BLOG , WORLD DRUG TRACKER
THANKS AND REGARD’S
DR ANTHONY MELVIN CRASTO Ph.D
DR ANTHONY MELVIN CRASTO Ph.D
MOBILE-+91 9323115463
GLENMARK SCIENTIST , INDIA
web link
web link
ABOUTME, BRAND ANTHONYCRASTO, COLLECTION OF SITE LINKS,BRAVESITES, GRAVATAR, JIMDO, SKILLPAGES, MIXXT, APNACIRCLE, ZIC ZAC, ZING ME, SCOOP IT, scribd, Stumbleupon, Delicious, pinnterest, tumblr, Newsvine, SLIDESHARE, ACADEMIA.EDU, GOOGLE PLUS, FACEBOOK, TWITTER, ISSUU, DIIGO,YOLASITE, Authorstream, Symbaloo, ISSUU, BLOGLOVIN, FRIENDFEED,NEWSVINE
Congratulations! Your presentation titled “Anthony Cra sto Glenmark scientist, helping millions with websites” has just crossed MILLION views.
アンソニー 安东尼 Энтони 안토니 أنتوني
blogs are 
SCALEUP OF DRUGS, ALL FOR DRUGS ON WEB,
MY CHINA, VIETNAM AND JAPAN BLOGS
ICELAND, RUSSIA, ARAB
GROUPS
you can post articles and will be administered by me on the google group which is very popular across the world
OPD GROUPSPACES, SCOOP OCI, organic-process-development GOOGLE, TVINX, MENDELEY WDT, SCIPEOPLE OPD,EPERNICUS OPD, SYNTHETIC ORGANIC CHEMISTRYLinkedIn group, DIIGO OPD, LINKEDIN OPD, WDT LINKEDIN, WDTI ZING
- drug regulatory affairs international
- ORGANIC SPECTROSCOPY INTERNATIONAL
- ORGANIC SYNTHESIS INTERNATIONAL
- ALL ABOUT DRUGS
- ORGANIC CHEMISTRY INTERNATIONAL
are some most read chemistry blogs, He has hands on experience in initiation and developing novel routes for drug molecules and implementation them on commercial scale over a 25 year tenure, good knowledge of IPM, GMP, Regulatory aspects, he has several international drug patents published worldwide .
He has good proficiency in Technology Transfer, Spectroscopy ,Stereochemistry , Synthesis, Reactions in Org Chem , Polymorphism, Pharmaceuticals , Medicinal chemistry , Organic chemistry literature ,Patent related site , Green chemistry , Reagents , R & D , Molecules ,Heterocyclic chem , Sourcing etc
He suffered a paralytic stroke in dec 2007 and is bound to a wheelchair, this seems to have injected feul in him to help chemists around the world, he is more active than before and is pushing boundaries, he has one lakh connections on all networking sites, He makes himself available to all, contact him on +91 9323115463, amcrasto@gmail.com
Products listed on our website are either in stock or can be resynthesized within a reasonable time frame. N-butyl,methylpyrrolidinium trifluoroacetate
ReplyDelete